三、中国口腔义齿行业发展环境分析
1.行业监管体制与法律法规
(1)国内口腔医疗器械行业监管体制
①分类管理制度
根据2021年6月1日施行的《医疗器械监督管理条例(2021修订)》规定:我国医疗器械产品的分类管理制度如下:第一类医疗器械:风险程度低,实行常规管理可以保证其安全、有效的医疗器械;第二类医疗器械:具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械;第三类医疗器械:具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械。
②医疗器械产品注册与备案制度
根据2021年6月1日施行的《医疗器械监督管理条例(2021修订)》和2021年10月1日施行的《医疗器械注册与备案管理办法》规定:第一类医疗器械实行产品备案管理,第二类、第三类医疗器械实行产品注册管理。第一类医疗器械产品备案,由备案人向所在地设区的市级人民政府负责药品监督管理的部门提交备案资料。申请第二类医疗器械产品注册,注册申请人应当向所在地省、自治区、直辖市人民政府药品监督管理部门提交注册申请资料。申请第三类医疗器械产品注册,注册申请人应当向国务院药品监督管理部门提交注册申请资料。
医疗器械产品注册、备案,应当进行临床评价。按照国务院药品监督管理部门的规定,进行医疗器械临床评价时,已有临床文献资料、临床数据不足以确认产品安全、有效的医疗器械,应当开展临床试验。开展医疗器械临床试验,应当按照医疗器械临床试验质量管理规范的要求,在具备相应条件的临床试验机构进行,并向临床试验申办者所在地省、自治区、直辖市人民政府药品监督管理部门备案。第三类医疗器械临床试验对人体具有较高风险的,应当经国务院药品监督管理部门批准。
③医疗器械生产许可与备案制度
根据2017年11月17日施行的《医疗器械经营监督管理办法(2017年修正)》规定:从事第一类医疗器械生产的,应当向所在地设区的市级人民政府负责药品监督管理的部门备案;从事第二类、第三类医疗器械生产的,应当向所在地省、自治区、直辖市人民政府药品监督管理部门申请生产许可。医疗器械生产许可证有效期为5年。
国务院药品监督管理部门负责制定医疗器械的分类规则和分类目录,并根据医疗器械生产、经营、使用情况,及时对医疗器械的风险变化进行分析、评价,对分类规则和分类目录进行调整。
④医疗器械经营许可与备案制度
根据2017年11月17日施行的《医疗器械经营监督管理办法(2017年修正)》规定:按照医疗器械风险程度,医疗器械经营实施分类管理。经营第一类医疗器械不需许可和备案,经营第二类医疗器械实行备案管理,经营第三类医疗器械实行许可管理。从事第三类医疗器械经营的,经营企业应当向所在地设区的市级食品药品监督管理部门提出申请,从事第二类医疗器械经营的,经营企业应当向所在地设区的市级食品药品监督管理部门备案。《医疗器械经营许可证》有效期为5年,《医疗器械经营许可证》有效期届满需要延续的,医疗器械经营企业应当在有效期届满6个月前,向原发证部门提出《医疗器械经营许可证》延续申请。
(2)海外口腔科用设备及器具制造行业监管体制
医疗器械出口国际市场的准入认可主要有欧盟CE认证和美国FDA注册等,具体如下:
①美国医疗器械监管体制
在美国,医疗器械的上市前审批由FDA统一管理。FDA对医疗器械实行分类管理,即根据风险等级和管理程度把医疗器械分为I、II、III三类,类别越高,风险越高。根据FDACFRTitle21,美国FDA市场准入的常规途径一般有三种:上市前通告(510K)豁免、PremarketNotification(PMN)即510K、PremarketApproval(PMA),具体如下:

数据整理:中商产业研究院
②欧盟医疗器械监管体制
欧盟将医疗器械分为4类,即I、IIa、IIb和III类,并规定对不同类别的医疗器械采用不同的审查方式,其中I类属于低风险医疗器械,由生产商自行评估是否符合MDR的相关规定并向生产所在国主管部门备案,IIa、IIb和III类医疗器械应当由公告机构进行符合性评估,通过评估后由公告机构签发认证证明,加贴CE标识。欧盟国家的“CE”认证属于强制性认证标志,产品需加贴“CE”标志才能在欧盟市场自由流通。
(3)行业相关产业政策
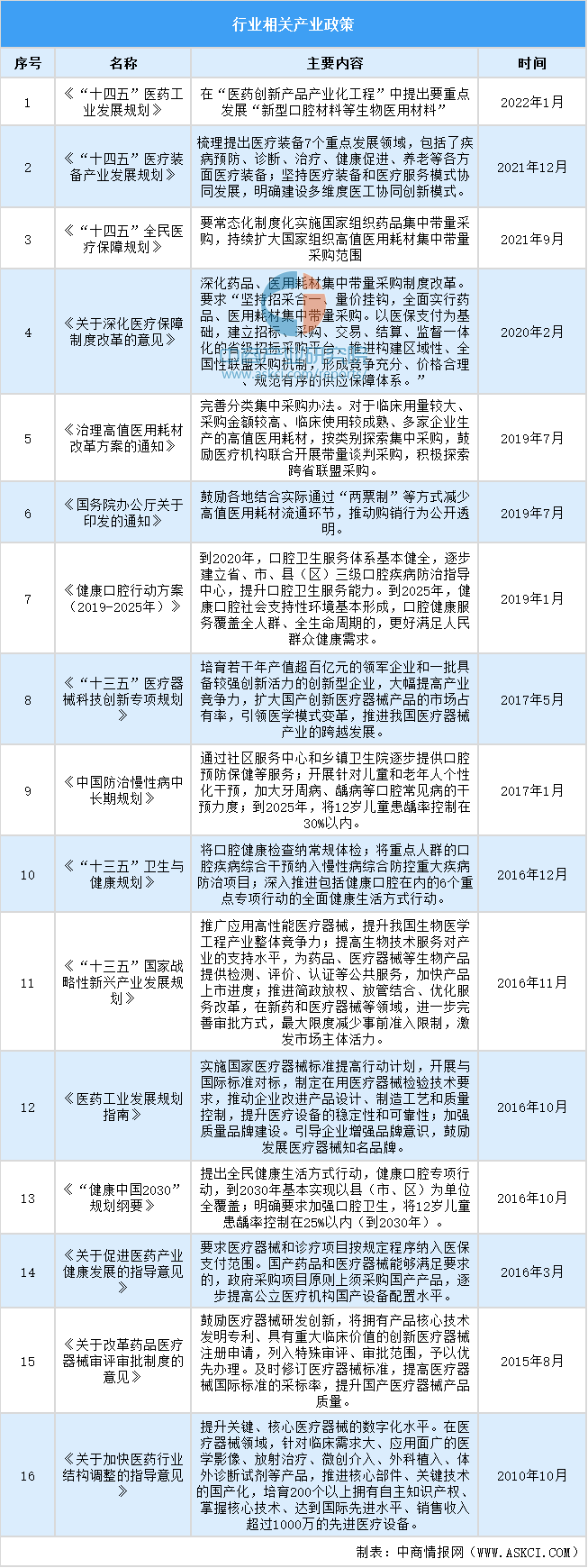
数据来源:中商产业研究院整理
 如发现本站文章存在版权问题,烦请联系editor@askci.com我们将及时沟通与处理。
如发现本站文章存在版权问题,烦请联系editor@askci.com我们将及时沟通与处理。


 2025年中国最受消费者喜爱的十大水果零售品牌排行榜(附榜单)
2025年中国最受消费者喜爱的十大水果零售品牌排行榜(附榜单)
 2025年中国最受消费者喜爱的十大月饼品牌排行榜(附榜单)
2025年中国最受消费者喜爱的十大月饼品牌排行榜(附榜单)
 2025年中国最受消费者喜爱的十大便利店品牌排行榜(附榜单)
2025年中国最受消费者喜爱的十大便利店品牌排行榜(附榜单)
 2025年1-7月中国新能源汽车销量前十企业(集团)排行榜(附榜单)
2025年1-7月中国汽车销量前十企业(集团)排行榜(附榜单)
中国工业机器人区域竞争力图谱:长三角全链领跑,珠三角场景突围(图)
2025年中国电源管理芯片产业链图谱及投资布局分析(附产业链全景图)
2025年中国精细化工行业市场前景预测研究报告(简版)
2025年7月中国煤及褐煤进口数据统计分析:进口量3560.9万吨
2025年7月中国粮食进口数据统计分析:进口量1405.7万吨
2025年1-7月中国新能源汽车销量前十企业(集团)排行榜(附榜单)
2025年1-7月中国汽车销量前十企业(集团)排行榜(附榜单)
中国工业机器人区域竞争力图谱:长三角全链领跑,珠三角场景突围(图)
2025年中国电源管理芯片产业链图谱及投资布局分析(附产业链全景图)
2025年中国精细化工行业市场前景预测研究报告(简版)
2025年7月中国煤及褐煤进口数据统计分析:进口量3560.9万吨
2025年7月中国粮食进口数据统计分析:进口量1405.7万吨